
Questi tre capitoli della norma sono strettamente connessi, ma non di rado vengono interpretati in modo un po’ confuso. Mi permetto, quindi, di (cercare di) gettare un po’ di luce.
Li considero però in un ordine che non è quello della norma, ma che a parer mio aiuta a capire meglio. Ed eviterò di entrare troppo nel dettaglio (c’è il testo della norma, per quello!), esaminandoli da un punto di vista operativo.
7.5.6: “Validazione dei processi di produzione ed erogazione dei servizi”
Questo capitolo si occupa di quelli che “ai miei tempi” venivano definiti processi speciali, ossia quei processi che (come recita la norma) “non possono essere verificati o non sono verificati mediante successivo monitoraggio o misurazione”, ad esempio perché l’attività di controllo sarebbe distruttiva del prodotto stesso.
Se non posso verificare il prodotto dopo averlo fabbricato, è logico che io debba assicurare di realizzarlo conforme in modo consistente, continuativo e coerente… cosa che è, appunto, l’oggetto della validazione.
La “Practical guide” della ISO riporta, in un’utile tabella, alcuni esempi di processi che dovrebbero richiedere o meno la validazione:
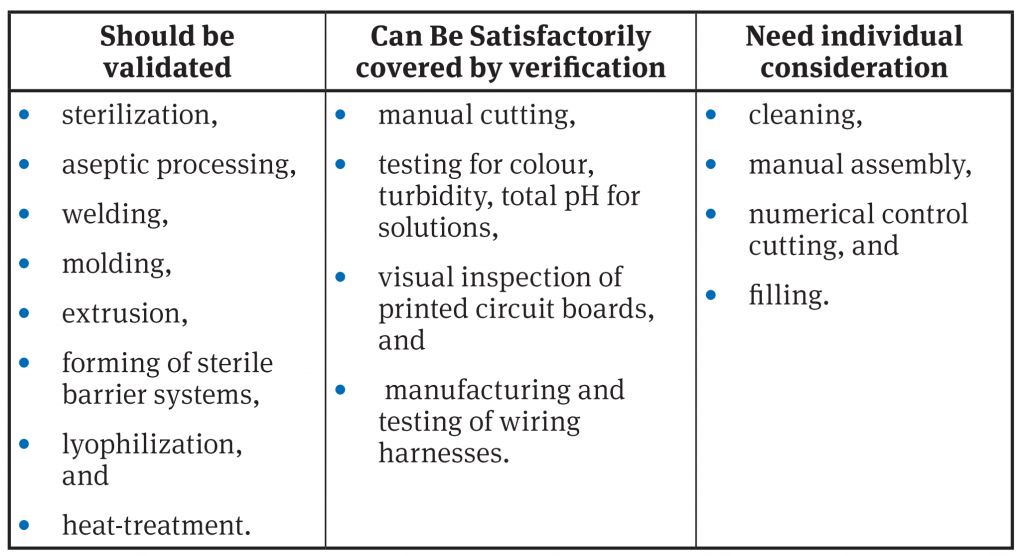
Ovviamente questa tabella va contestualizzata!, anche considerando i rischi associati a quel determinato processo.
Un tipico esempio di processo speciale, è proprio la sterilizzazione: il test di sterilità sul campione, infatti, lo rende inutilizzabile. Stessa cosa per il processo di formazione del packaging sterile, laddove – per verificare l’integrità del packaging – sono costretto a violarlo.
In questi due casi, però, la criticità è tale che la ISO 13485 riserva un capitolo a parte: il 7.5.7.
Come auditor, nella nostra checklist del punto 7.5.6 andremo quindi a trattare gli altri processi speciali (ed anche il software) che l’azienda dovesse mettere in atto in produzione, e fossero inclusi nel campo di applicazione del certificato.
INCISO: confesso di non amare la parola “convalida”! Io preferisco usare solo questi due termini:
- Validazione: si applica a oggetti non materiali, come ad esempio i processi, o il software,
- Qualifica: si applica a oggetti materiali, come ad esempio attrezzature, sistemi, hardware, ambienti a contaminazione controllata, eccetera. La qualifica può essere parte della validazione (vedi IQ, OQ, PQ).
7.5.7: “Requisiti particolari per la validazione dei processi per la sterilizzazione e i sistemi di barriera sterile”
Lo possiamo considerare un sotto-capitolo del 7.5.6, per i motivi spiegati sopra.
Che cos’è un sistema di barriera sterile? La domanda non è scontata… Specie per chi è uso ai dispositivi non sterili! Ecco quindi la definizione della norma che se ne occupa, la EN ISO 11607-1:2020 (traduzione mia):
Imballaggio minimo che minimizza il rischio di ingresso di micro-organismi, e consente una presentazione asettica del contenuto sterile nel punto di utilizzo.
Per soddisfare il punto norma 7.5.7, quindi, occorre avere evidenze oggettive relative alla documentazione (protocollo, report, e relativi allegati) di validazione, per questi due processi: sterilizzazione, e formazione del sistema di barriera sterile.
Nel rispondere ai requisiti di questo capitolo, entrano necessariamente in gioco le varie norme specifiche… come quella sopra citata, oppure la EN ISO 11135:2014 nel caso del processo di sterilizzazione con Ossido di Etilene.
7.5.5: “Requisiti particolari per dispositivi medici sterili”
Questo capitolo, personalmente, lo vedo come un sotto-capitolo del 7.5.1: la raccolta, ad esempio, delle evidenze relative al ciclo di sterilizzazione per il batch record che è stato campionato come rappresentativo della produzione.
Infatti, nella mia lista di riscontro, io di solito inserisco tutte queste evidenze nel 7.5.1; e poi, nel 7.5.5, faccio riferimento a quest’ultimo punto.




